Posted on : October 21st 2024
Regulatory submission documents in pharmaceutical R&D are comprehensive dossiers that present detailed information about a drug candidate to regulatory authorities. These documents encompass a wide range of data, including preclinical studies, clinical trial results, manufacturing processes, and quality control measures. The content and format of these submissions are typically standardized to facilitate efficient review by regulatory bodies such as the FDA in the United States or the EMA in Europe.
The specific documents required can vary based on the stage of drug development and the type of submission. For example, an Investigational New Drug (IND) application includes information on the drug’s composition, manufacturing, and preclinical data to support initial clinical trials. In contrast, a New Drug Application (NDA) or Marketing Authorization Application (MAA) provides a complete picture of the drug’s development journey, including all clinical data, proposed labeling, and risk management plans. These documents serve as the foundation for regulatory decision-making, determining whether a drug can proceed to clinical trials or be approved for market release.
Some of these documents include:
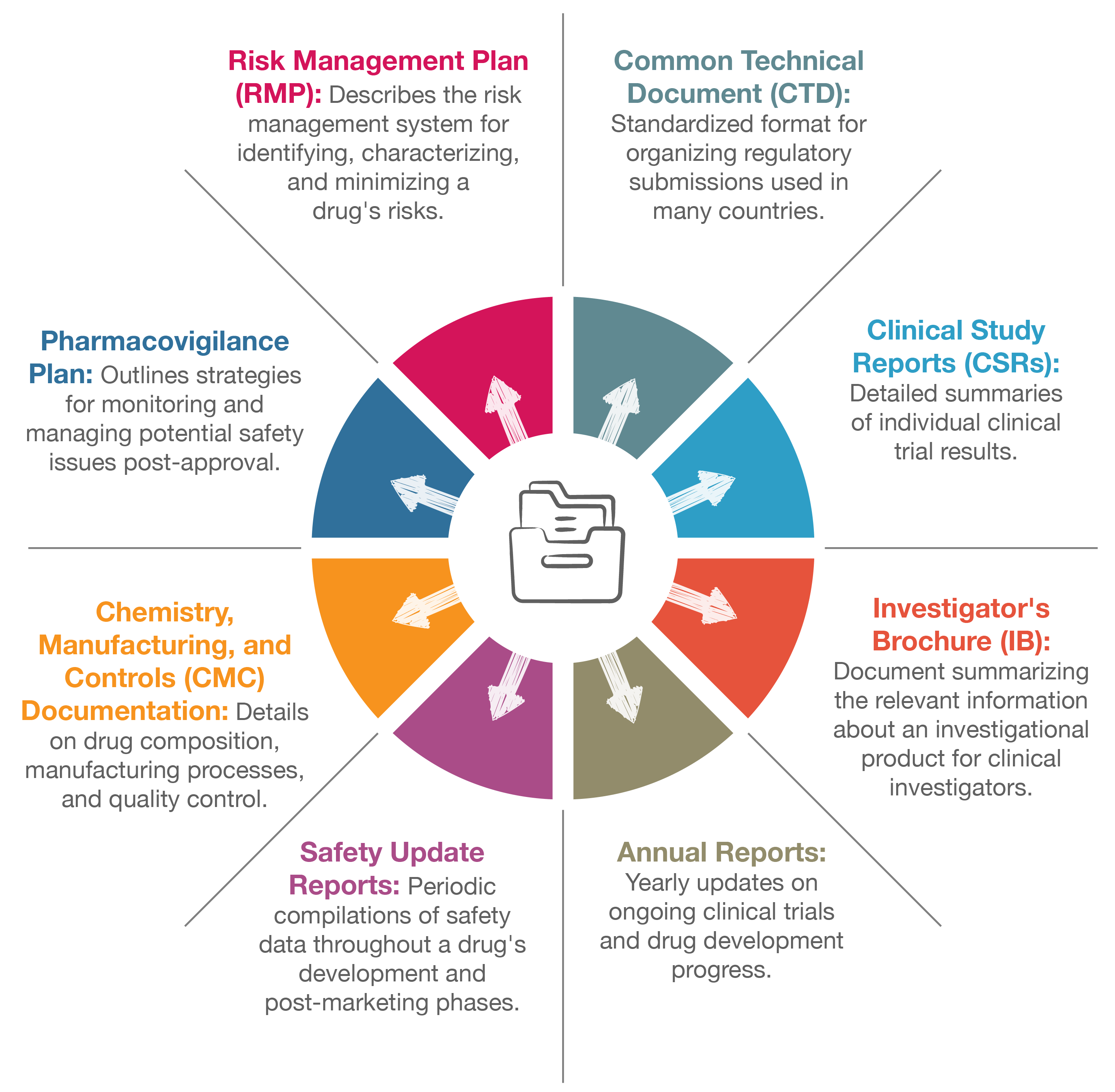
Each region, such as the U.S. (FDA), the European Union (EMA), and other jurisdictions, has specific requirements regarding the content and structure of these submissions, adding an additional layer of complexity to the process. For global pharmaceutical companies, complying with various regional regulations is critical to achieving market authorization in multiple countries.
Read more – Advanced Analytics for Pharmaceutical Regulatory Workflows
The Lifecycle of Regulatory Submissions
The lifecycle of regulatory submissions in pharma R&D spans several stages, beginning with the collection of clinical trial data and culminating in the final submission to regulatory authorities. Each stage is critical to ensuring that all required information is accurately compiled and presented in a way that meets regulatory standards.
- Data Collection: The process begins with gathering data from preclinical studies and clinical trials. This data includes safety, efficacy, pharmacokinetics, and pharmacodynamics information, all of which will form the foundation of the submission. Multiple sources, including trial sites, real-world evidence, and laboratory studies, contribute to this pool of data.
- Data Analysis and Validation: Once the data is collected, it is analyzed to assess the drug’s safety and efficacy. Cross-functional teams, including clinical development, biostatistics, and pharmacovigilance teams, evaluate the trial outcomes and perform rigorous checks to ensure that the data is valid, accurate, and consistent. The findings from these analyses are then structured into specific regulatory documents like the ISS and ISE.
- Content Generation and Review: This is one of the most labor-intensive stages, where teams begin drafting key documents such as Clinical Study Reports (CSRs), clinical protocols, and integrated summaries. Regulatory experts, medical writers, and statisticians play a significant role in generating narrative sections, interpreting data, and ensuring that the content aligns with both internal standards and external regulatory requirements.
- Submission Preparation: Once the documents are drafted, they undergo multiple rounds of internal reviews and revisions. Legal and compliance teams often step in to verify that the content adheres to regulatory guidelines, while senior management provides final approval before the submission is packaged for delivery.
- Final Submission to Regulatory Authorities: After all documents are reviewed and approved, they are formally submitted to regulatory agencies such as the FDA or EMA. These agencies conduct their own review process, and further requests for clarification or additional data may arise, which requires ongoing coordination among the teams involved.
Throughout this lifecycle, cross-functional collaboration is paramount. Teams from different domains—clinical, regulatory, medical writing, and biostatistics—must work seamlessly to produce cohesive, compliant, and accurate submissions. Miscommunication or delays at any stage can lead to significant bottlenecks, impacting timelines and ultimately delaying the approval process.
Current Challenges in Regulatory Submissions
The process of generating regulatory submissions for new drugs is fraught with challenges that affect both the speed and accuracy of the submission. These challenges stem from several critical pain points in data management, content generation, compliance, and team coordination.
- Data Complexity: The growing complexity of clinical trial designs, coupled with the need to incorporate real-world evidence, presents one of the biggest challenges. Modern clinical trials often involve multiple treatment arms, global trial sites, and diverse patient populations. This results in vast, fragmented datasets that must be synthesized into a coherent narrative for regulators. Furthermore, evolving safety reporting requirements add to this complexity, demanding rigorous validation and cross-checking of data from different sources. Manually managing these vast datasets can lead to errors, misinterpretations, and inconsistencies in the final submission, which increases the likelihood of regulatory pushback.
- Content Generation: Drafting the core documents of regulatory submissions—such as clinical protocols, safety reports, and efficacy summaries—is an immensely time-consuming and manual process. This requires expertise from multiple domains, including regulatory affairs, medical writing, and clinical teams. Each document must adhere to specific formatting and content guidelines, depending on the regulatory body. The iterative nature of this process, where drafts undergo several rounds of internal reviews, introduces the potential for human error and inconsistencies. This manual effort often leads to bottlenecks, as content generation becomes a time sink, especially when multiple regulatory filings are prepared in parallel.
- Regulatory Compliance: Navigating the regulatory landscape is becoming increasingly complex, with varying requirements from different jurisdictions (e.g., FDA, EMA, PMDA). Regulatory bodies may have different standards for the presentation of safety data, the inclusion of real-world evidence, or how efficacy is measured. Companies must ensure that their submissions are tailored to meet each authority’s specific criteria, adding another layer of complexity to the content generation process. Non-compliance or errors in meeting these diverse regulatory requirements can result in submissions being delayed or rejected.
- Version Control and Collaboration: With multiple cross-functional teams working on different aspects of the submission, managing version control is a major challenge. Different teams may be using different versions of data or documents, which can lead to inconsistencies in the final submission. Keeping track of updates and ensuring that all stakeholders are working from the most current version of a document is a difficult task, especially in large, global pharmaceutical organizations. The lack of a centralized system to monitor changes across all documents and datasets can result in miscommunication and duplicated efforts.
- Timelines and Costs: Delays in the submission process are not uncommon, and they can have a significant impact on both timelines and costs. Manual processes, such as drafting, reviewing, and editing submission documents, are prone to delays due to the sheer volume of work involved. Missed deadlines not only delay the drug approval process but also increase costs due to extended resource allocation, rework, and the potential for missed market opportunities. For pharmaceutical companies, reducing the time to submission is critical to maximizing the commercial window of a new drug, but the current manual-heavy approach often impedes timely submissions.
The Need for Innovation
The current regulatory submission process in pharma R&D is weighed down by significant inefficiencies and complexities. From managing vast datasets and ensuring regulatory compliance across multiple jurisdictions to manually drafting and reviewing intricate documents, the challenges faced by cross-functional teams can slow down timelines and increase the risk of costly delays. These hurdles make it difficult for pharmaceutical companies to efficiently bring new therapies to market, often creating bottlenecks at critical stages of the drug approval process.
To address these challenges, the industry must look toward innovative solutions like Generative AI. By leveraging AI-driven tools, companies can streamline content generation, automate data integration, and reduce manual errors. Generative AI has the potential to enhance efficiency and quality of regulatory submissions by accelerating the drafting process, ensuring compliance with evolving regulatory requirements, and enhancing collaboration across teams.
In the next part of this series, we will explore how a GenAI-based tool can specifically address these pain points and enhance the way regulatory submissions are created and managed.
About the Author

Santosh Shevade is a Principal Data Consultant at Gramener – A Straive Company. With deep expertise in healthcare strategy, digital health, and clinical development and operations, he has supported nearly 50 clinical development programs across all clinical phases. His experience spans advanced analytics solution design for pharmaceutical companies, mHealth implementation, and AI applications in healthcare. Previously at Novartis and Johnson & Johnson, Santosh led global clinical development teams and streamlined data review processes for major regulatory submissions. A certified MBTI trainer and leadership coach, he serves as visiting faculty at ISB Hyderabad and Welingkar Institute, focusing on healthcare technology innovation and biopharma strategy.